立体構造予測
「タンパク質の立体構造をコンピュータで予測する」
■背景 タンパク質の立体構造を知ることは、タンパク質の機能を理解し、生物のしくみを理解するのに重要な手がかりとなります。現在、さまざまな生物のゲノムの解読が進んでいますが、遺伝子から翻訳されるタンパク質で、立体構造がわかっていないタンパク質は、たくさんあります。このため、時間と労力をあまりかけない、コンピュータを使ったタンパク質の立体構造予測が期待されています。私たちは、データベースに蓄積された情報と、物理に基づく解析の両方から、タンパク質の立体構造(以下では単に構造と呼びます)を高い精度で予測する研究に取り組んでいます。
■研究概要
タンパク質の構造を予測する一番ポピュラーの方法は、データベースに登録されている、すでにわかっているタンパク質の構造の中から手本(鋳型)となる構造を選んで予測構造を作るという方法です。良い手本を得るには、構造がわからないタンパク質とアミノ酸の並びが似ているものを探すという方法がとられますが、私たちは、そうしたタンパク質を多数集めて、その特徴を統計的に解析し、どこをどのように手本とすればよいかを決める方法について研究しています。また、手本となる構造が見つからなくても、参考になりそうな構造のパーツを組み上げて予測構造を作る方法も開発してきました。さらに、予測した構造のクオリティを、データベースに登録されているたくさんのタンパク質の構造を使ってコンピュータにその特徴を学習させ、評価する方法も開発しています。
分子シミュレーションは、原子間に働く力をもとに原子の動きを計算する方法で、いろいろなところで応用されていますが、私たちは、溶媒の存在を考慮した分子シミュレーションを使って、生体内で特定の構造に折りたたまれるフォールディングの過程を解析する研究と、上で述べたような方法で作った予測構造からシミュレーションをスタートさせ、構造を修正して、予測の精度をさらに高める研究を行っています(図1、文献1)。
また、タンパク質とタンパク質がどのように結合しているか、つまりタンパク質とタンパク質の複合体の構造を予測するドッキング予測の方法も開発しています。ドッキング予測では、あらゆる方向から、あらゆる角度で2つの分子をくっつけてみるということをしますが、そのための高速計算アルゴリズムを新たに開発して、従来よりも16~160倍の高速化を実現しました(図2、文献2)。
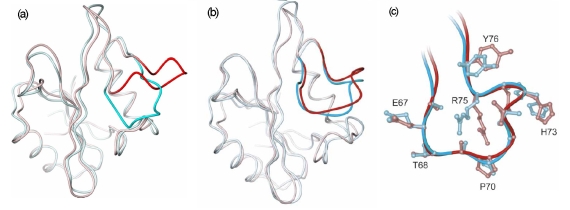
図1 精密な構造予測の例
(a)は、血清アミロイドP成分(SAP)タンパク質のSH2ドメインの構造を、チロシンキナーゼp56lckのSH2ドメインの構造を手本(鋳型)にして予測した結果を示しています。SAPタンパク質の結晶構造はすでに得られていますが、それがわからないと仮定して予測しています。青色は結晶構造、赤色は予測構造です。色が濃い部分は、予測構造を作る際、対応する鋳型の構造が得られなかった部分で、予測構造が結晶構造と大きくずれているのがわかります。(b)は、(a)の結晶構造と予測構造からスタートしてシミュレーションを行い、それぞれのシミュレーションでよく現れる代表的な構造を示したものです(色が対応しています)。シミュレーションにより、予測構造が結晶構造に近づいていること、またその構造は溶媒中で安定した構造をとっていることがうかがえます。(c)は、(b)の図を拡大した図です。
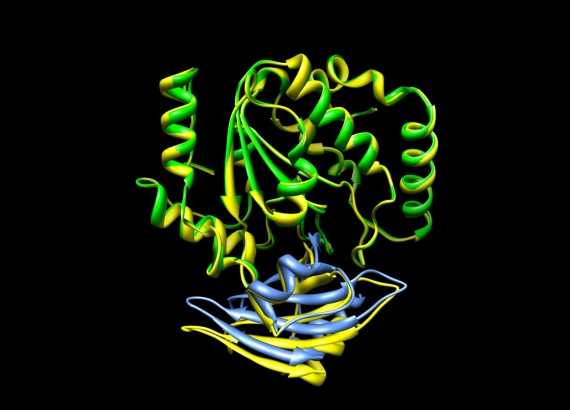
図2 ドッキング予測の例
グリコシラーゼ(緑色)とそのインヒビター(青色)とのドッキング予測の結果です。黄色は、すでにわかっている複合体の結晶構造で、予測した結果と重ね合わせて表示しています(角越和也助教による)。
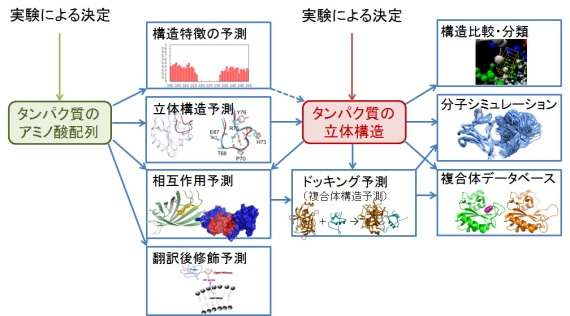
図3 タンパク質のさまざまな予測・解析ソフトウェアの開発
■科学的・社会的意義 精度の高いタンパク質の立体構造予測は、生物のしくみの解明、薬剤や酵素の設計、食品の開発などで基盤となる重要な研究です。私たちは、タンパク質の立体構造予測のほか、コンピュータを使ったタンパク質のさまざまな予測・解析ソフトウェアを開発していて(図3)、ライフサイエンスの研究者と多くの共同研究を行っています。
■参考文献
1) R. Ishitani, T. Terada, K. Shimizu (2008). “Refinement of comparative models of protein structure by using multicanonical molecular dynamics simulations,” Molecular Simulation, 34, 327-336.
2) K. Sumikoshi, T. Terada, S. Nakamura, K. Shimizu (2011). “Protein-Protein Docking Using Multi-layered Spherical Basis Functions,” Proceedings of the 2011 International Conference on Bioscience, Biochemistry and Bioinformatics, 342-347.
 H25年度分野別専門委員
H25年度分野別専門委員
東京大学・生物情報工学
清水謙多郎 (しみずけんたろう)
https://www.bi.a.u-tokyo.ac.jp/~shimizu/
「全原子シミュレーションによる蛋白質のフォールディング問題」
■背景
蛋白質は20種類からなるアミノ酸がペプチド結合をしてつながった1本鎖の高分子化合物です。構成するアミノ酸の数や種類や配列(1次配列)により、生理的な条件下で蛋白質の立体構造(3次構造)が決まります。アミノ酸の配列は、遺伝子(DNA)の塩基配列により決まります。現在、ゲノムの解析によりアミノ酸配列が分かっているのに対し、立体構造は分かっていない蛋白質がたくさんあります。蛋白質の立体構造が分かれば、どのように他の蛋白質と結合するか、どう構造変化を起こすか、また、機能について理解しやすくなります(図1)。立体構造予測とは、アミノ酸配列の情報から蛋白質の未知の立体構造を予測することです。
計算機を用いた立体構造予測には、データベースに基づいたものと化学物理に基づいたものがあります。データベースに基づいたものは、すでに配列と構造が分かっている蛋白質のデータベースを構築し、このデータベースを使って構造予測します。(既存の配列と似た1次配列の部分は、その配列に対応する既存の立体構造に似ているとして、立体構造予測を行います。)化学物理に基づいたものは、原子間やアミノ酸残基間のエネルギーや作用する力を化学物理に基づいて定義し、安定な構造(多次元空間の自由エネルギーが最小になる状態)を探します。(いろいろな空間・時間スケールのものがあり、原子にかかる力を定義して、運動方程式を解いて安定な構造を予測するものもあれば、粗視化をおこないアミノ酸残基を1つのサイトと考え、サイト間のエネルギーを定義して、エネルギーの安定な構造を予測するものもあります。)
データベースを使った方法は強力な方法ですが、データベースにない配列や、蛋白質のまわりの溶媒条件などを変えた場合の構造変化には、化学物理に基づいた方法が威力を発揮します。ここでは、物理化学に基づいた全原子シミュレーションによる蛋白質のフォールディング問題について少し説明します。
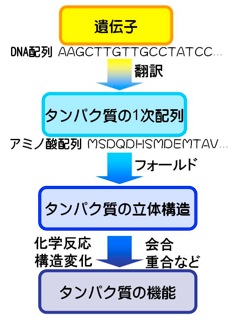
図1 DNA配列→アミノ酸配列→立体構造→機能
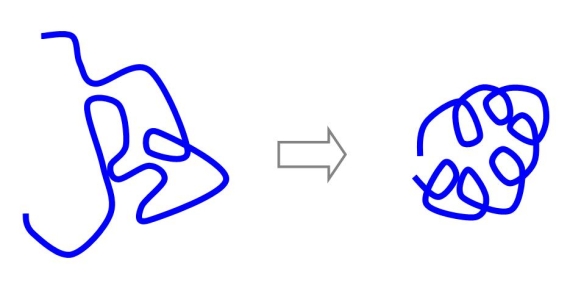
図2 フォールディングの概要図
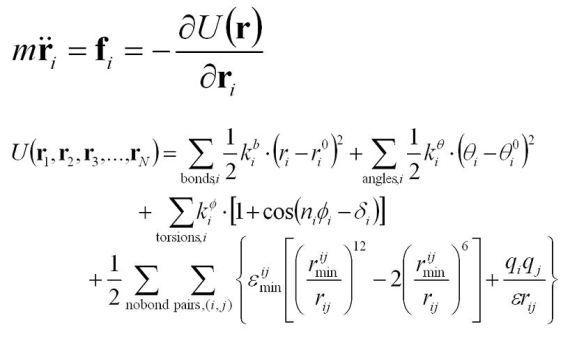
図3 運動方程式と蛋白質のポテンシャルエネルギー関数
 | 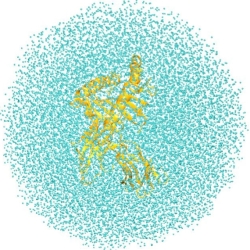 |
図4 蛋白質と周りの水分子(左図は蛋白質を原子表示、右図は蛋白質をリボン表示した。水色の粒子は水分子を表す。)。蛋白質の周りの溶媒分子は蛋白質の構造や機能に大きな影響を与える。 (描写にはVMDとraster3Dを使用)
■研究概要
蛋白質は、生理的条件では1つの立体構造を持ちます。100残基程度の蛋白質には溶媒環境を変えたり、高温にすると立体構造が壊れますが、生理的条件にもどすと、もとの構造にもどるものがあります。蛋白質がもとの構造に戻ることをフォールディング(巻き戻り)といいます(図2)。大体、ms程度の時間がかかります。
全原子シミュレーションでは、ポテンシャルエネルギー関数に基づいて原子間に作用する力(詳しく述べませんが、ポテンシャルエネルギー関数の傾きが力になります。)を計算し、ニュートンの運動方程式を用いて、座標の時間変化を追います(図3)。たくさんの原子間の力を計算するので計算機が必要です。大体、原子の空間スケールが0.1nm程度で時間変化の1ステップが数fs(10-15秒)になります。現在の計算機ではms(10-3秒)程度のシミュレーションを行うことは困難です。(系を固定してハードウェアーから設計してms程度のシミュレーションを実現しようとしている研究グループもあります。)
フォールディングするのにms程度かかるのは、いろいろな構造の中から自由エネルギーが最小な安定構造を探すのに時間がかかるからと考えられています。シミュレーションでは、いろいろな状態から最安定状態を見つけるのはとても難しい問題(サンプリング問題)です。このサンプリング問題を解決するためには、化学物理の手法を駆使した効率良く系の状態をサンプリングするサンプリング手法の開発が必要です。また、蛋白質のシミュレーションを行う場合は、先に述べたポテンシャルエネルギー関数が必要です。蛋白質やまわりの溶媒の原子間の力は1960年ごろから長い歴史の間に開発されてきましたが、まだ不十分なところもあります。また、特に蛋白質の周りにたくさんの溶媒分子がいて、蛋白質の揺らぎや機能に大きく影響を与えます(図4)。溶媒の効果を見積もるために蛋白質の周りに実際に水分子をおいてシミュレーションすることが多いのですが、これには大変計算時間がかかります。水の効果を取り入れる手法の開発が必要です。
これらの問題のため、全原子シミュレーションにより伸びた構造から蛋白質のフォールディングを実現するのは難しいです。これまで、フォールディング問題のために、化学物理の理論に基づいた強力なサンプリング手法の開発や溶媒効果を効率良くとりいれることを行ってきました。化学物理の手法に基づいた全原子シミュレーションを駆使して、ランダムな構造からアミノ酸配列のみの情報で蛋白質の立体構造を予測することは大変ですが、面白い研究分野です。(近年では、これらの手法を駆使し、また計算機パワーの向上により、全原子シミュレーションにより数十残基の蛋白質のフォールディングが実現可能になってきています。)
■科学的・社会的意義 サンプリング手法の開発、力場の改良により、全原子シミュレーションから蛋白質の立体構造を予測できれば、計算機で蛋白質の構造や機能について原子レベルで研究できるようになり非常に重要です。たとえば、一部のアミノ酸配列を変えることによって、機能がかわる蛋白質などもありますが、これらの機構を解明することができます。また、蛋白質の会合などについての機構も解明でき、創薬などの応用分野にも使えます。
H24年度分野別専門委員
慶應義塾大学・理工学部・物理学科
光武亜代理 (みつたけあより)
https://www.phys.keio.ac.jp/faculty/ayori/syousai/